Was ist PRA?
Die progressive Retinaatrophie (PRA) ist ein Überbegriff für eine Gruppe erblicher Netzhauterkrankungen, die auf degenerativen oder dysplastischen (unterentwickelten) Veränderungen der lichtempfindlichen Fotorezeptoren (Stäbchen, Zapfen) in der Netzhaut basieren.
Als Ursache sind unterschiedliche Gendefekte (Mutationen) bekannt, die zu unterschiedlichen Störungen auf zellulärer und molekularer Ebene in der Netzhaut führen. Dementsprechend hat man „Unterbezeichnungen“ (z. B. prcd, rcd1, rcd2, rcd3, crd1, crd2, XL PRA) für die einzelnen PRA-Formen eingeführt.
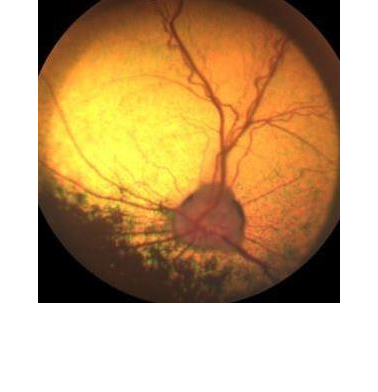
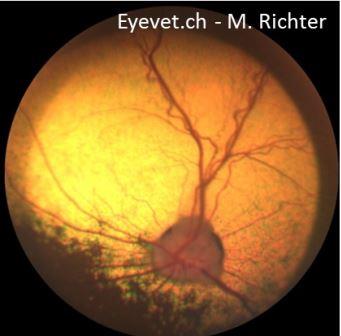
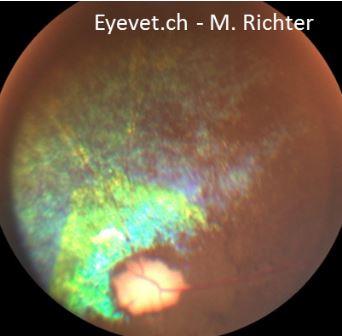
Das Endresultat ist jedoch bei fast allen PRA-Formen gleich: totale Atrophie der Netzhaut. Im Verlauf der PRA wird die gesamte Netzhaut dünner (man spricht von Atrophie) und bei der Untersuchung des Augenhintergrundes bzw. der Netzhaut erkennt man eine Atrophie der Netzhautgefässe, ein verstärktes „Leuchten“ des Tapetum Lucidum (welches direkt unterhalb der zunehmend dünner werdenden Netzhaut liegt und somit einfallendes Licht verstärkt reflektiert), schollenartige Pigmentverschiebungen im Bereich des nicht-tapetalen Fundus und schlussendlich auch eine Atrophie des Sehnervenkopfes.
Die PRA tritt in beiden Augen symmetrisch auf. PRA beim Hund ist vergleichbar mit der erblichen Retinitis pigmentosa des Menschen.